





CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)技术自2013年被科学家首次证明可以在哺乳动物细胞内实现高效的基因组编辑以来,正在彻底改变生物医学基础研究和临床研究领域。越来越多的研究者已经在包括开发新型基因编辑工具酶或者优化向导RNA(gRNA)的设计方面进行了很多尝试,希望可以借此提高CRISPR的性能。
利用合成gRNA-靶序列的高通量文库允许直接在细胞环境中便捷和高通量地收集gRNA活性数据,由此建立的计算模型来预测gRNA的活性比较可靠。在以往的研究中,约10000至50000条合成的gRNA-靶序列被用于量化gRNA的靶向效率、特异性或修复结果。然而,人类基因组有6X108个具有NGG PAM的潜在gRNA,这导致早期研究中采样的gRNA覆盖率仅为0.002%——0.009%。
在这个采样范围内,已有的计算工具预测的gRNA活性和用于建模的实测gRNA活性相关性约为0.8(Spearman相关性),还有很大上升空间。并且截至目前,还没有一个从多个维度预测gRNA活性(如:gRNA切割活性、gRNA脱靶活性、gRNA切割后基因组修复的图谱)的综合工具。
2023年5月16日,西湖大学生命科学学院马丽佳研究员和团队成员们在 Cell Discovery 上发表研究论文Deep sampling of gRNA in the human genome and deep-learning-informed prediction of gRNA activities。该论文介绍了团队开发的一种基于全新策略构建的深度学习模型,能有效预测CRISPR多维gRNA性能。论文链接:https://www.nature.com/articles/s41421-023-00549-9
首先,研究人员将74万条gRNA(740k文库)及gRNA靶点序列合成在一条oligo上(每个gRNA靶点序列包括20-nt上游基因组序列、20-nt靶标序列、3-nt PAM序列和20-nt下游基因组序列),该文库除了团队设计的gRNA之外,还包含多个已发表的CRISPR KO screening(通过CRISPR系统构建基因组文库进行高通量基因敲除筛选)文库,包括Brunello、GecKOv2、Sabatini、TorontoKoV3和YusaKoV1。
740k文库占人类基因组中所有具有NGG-PAM的gRNA数量的约0.16%(远高于以往研究中0.002%——0.009%的覆盖率)。随后通过慢病毒转导入稳定表达SpCas9的人体细胞系内,对合成的gRNA靶点序列上下游设计引物PCR进行深度测序即可实现高通量地检测gRNA的活性,同时还可评估对应gRNA切割基因组后,基因组的修复图谱(图1)。
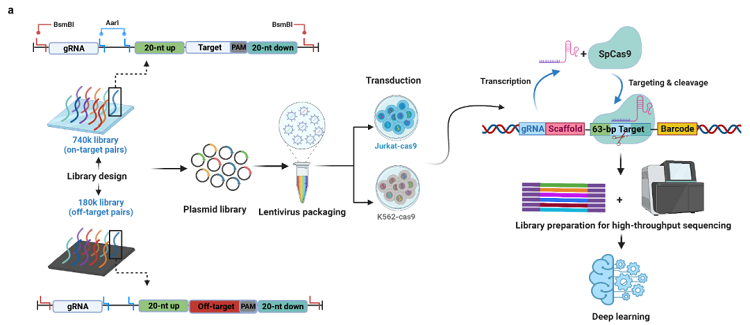
图1 高通量检测gRNA体内活性策略的流程图
为了开发gRNA活性预测模型,研究人员首先比较了九种机器学习算法,包括七种传统算法和两种基于深度学习的算法,结果发现RNN是在所有测试算法中性能最好,实测的gRNA效率和预测效率之间的Spearman相关系数在0.875到0.911之间。值得注意的是,相比这项研究中使用了74万条gRNA,以往的研究只用了1万至5万条gRNA序列的数据进行深度学习模型构建。
那么,在数据量和模型性能之间,什么数据量是生物学实验投入和模型性能获益之间的最佳值呢?带着这个疑问,研究人员又将740k文库分为8个子库,分别叠加子文库行深度模型构建,利用十折交叉验证进行评估,最终建立了8组模型,结果表明随着文库大小的增加,8组模型的预测性能Spearman相关系数中位数从0.810逐渐提高到0.898。在包含超过220k个gRNA后,模型性能趋于平稳。
本研究给出了gRNA数据规模对提升模型性能的重要参考,对后续类似工作如何设计实验具有重要的指导意义。随着一系列建模参数的测试,研究人员最终确定将63bp的序列(23bp靶序列和上游、下游各20bp)作为RNN模型的输入序列,并将其训练得到的深度学习模型命名为“AIdit_ON”(图2)。
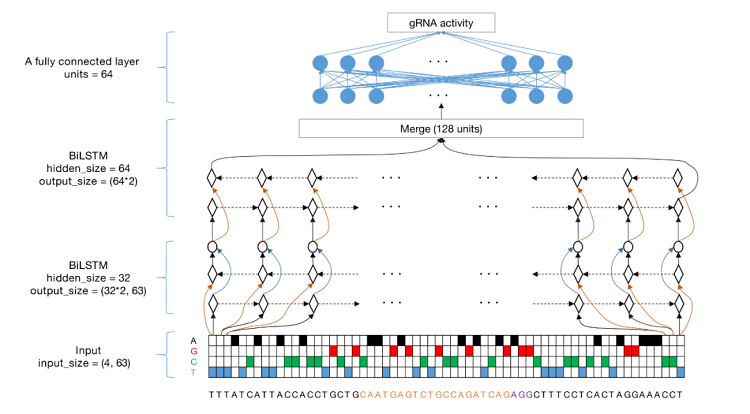
图2 建立预测gRNA活性的AIdit_ON模型的流程图
为了进一步评估AIdit_ON模型在不同细胞类型中的泛化性能,研究人员基于多个公开可用的内源数据集,比较了AIdit_ON和11个已发表的计算工具,结果显示AIdit_ON模型在所有公共数据集中的表现均要优于所有其他模型(图3)。同样地,对于本研究产出的不同细胞系内源的gRNA活性数据(293T n=78;K562 n=75;H1 n=71),AIdit_ON模型的表现也优于其他模型(图3)。
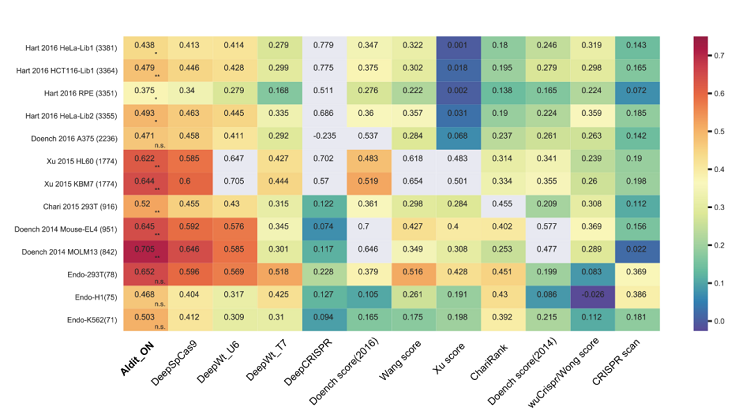
图3 不同数据集的indel频率和12个模型预测效率之间的Spearman相关系数的热图
此外,K562和Jurkat细胞中740k文库的数据集使研究人员能够进一步检测这两种细胞类型中DSB(DNA双链断裂)诱导的基因组修复结果。在仔细分析Jurkat和K562细胞的DSB修复图谱时,研究人员有了意外的发现。在K562中,超过65%的SpCas9/gRNA诱导的DSBs被修复为缺失类型(Deletion),这与以往研究的大多数细胞类型中发现的现象一致。
相反,插入(Insertion)在Jurkat细胞中占主导地位(大于60%)(图4)。研究人员大胆假设,Jurkat细胞的一种独特特性可能是导致Insertion主导修复结果的原因。经过验证,研究人员证实了他们的猜想,他们找到了一个在Jurkat特异性高表达的基因DNTT,该基因是控制Jurkat中DSB修复的主导基因。敲除Jurkat中的DNTT会改变其修复结果,使其与K562细胞的修复结果更相似。相反,在K562细胞中过表达DNTT时,Insertion成为最主要的DSB修复图谱。这些数据表明,由SpCas9/gRNA介导的DSB诱导的修复结果是DNTT依赖性的。
为构建精准预测DSB修复图谱模型,研究人员首先为每个修复类别训练了一个单独的XGBoost模型,这些模型的输出结合序列特征和微同源特征成为最终模型训练的特征。在K562数据上训练的模型被命名为AIdit_DSB_K562,在Jurkat数据上训练过的模型被称为AIdit_DSB_Jurkat。
为了评估模型的泛化性能,研究人员比较了已公开发表的预测模型ForeCasT和Lindel。在所有非Jurkat细胞系的测试数据集中,AIdit_DSB_K562模型的表现优于Lindel和ForeCasT模型。基于从不同系收集的其他公共数据集的结果,包括REP1、CHO、E14TG2A和HAP1,也证明了AIdit_DSB_K562模型的优越性能。
AIdit_DSB_Jurkat对非Jurkat数据的预测效力低,但在Jurkat测试数据集中实现了高精准的预测。根据DSB修复的DNTT依赖性,后续研究人员可以用DNTT的表达量来指导模型选择和预测不同细胞类型特异的SpCas9/gRNA DSB修复图谱。
这个重要现象的发现,解释了有些情况下模型预测性能无法在不同细胞类型中实现泛化的原因,其实是背后的基本生物学原理决定的。这也提示生物学数据指导下的AI模型,既需要产生足够的、与问题直接相关的数据,也需要考量有生物学意义的参数。

图4 SpCas9诱导的DSB修复类型在Jurkat WT、 Jurkat DNTT-KO、K562 WT和K562 DNTT-OE细胞中的分布
最后,研究人员应用了类似的策略来衡量SpCas9/gRNA在非匹配靶序列上的脱靶活性。研究人员设计了一个包含180万条gRNA脱靶序列的文库(180k),并基于该文库产生的高通量测序数据构建了预测SpCas9/gRNA脱靶活性的机器学习模型,即AIdit_OFF。
结果表明,在多个GUIDE-seq测试数据集上相对于应用较广的CFD模型,AIdit_OFF无论在特异性还是召回率上都表现得更好,且将预测gRNA脱靶位点的精准率平均提高了2.6倍。
为了方便生物医学研究人员更好选择gRNA,本项目搭建了一个嵌入三个高性能模型的公共网站(https://crispr-aidit.com)。研究者通过输入基因名称、序列片段或带有序列的FASTA文件,可以获得gRNA的多维预测数据,更精准选择合适的gRNA。