





中山大学药学院李民课题组近日在权威期刊Redox Biology发表题为“Suppression of ATG4B by copper inhibits autophagy and involves in Mallory body formation”的研究论文,揭示了铜离子通过抑制ATG4B导致自噬失活,进而阻碍威尔森病蛋白聚集体清除的一种新机制。
铜离子是人体必需的微量元素,但其过量蓄积会导致疾病发生,如威尔森病。威尔森病是ATP7B基因突变导致的常染色体隐性遗传病,ATP7B基因突变使其编码的ATP7B蛋白功能受损,肝细胞胆管侧的ATP7B无法转运铜离子至胆汁以排泄,过量蓄积的铜离子经肝细胞血管测进入血液循环,引起全身多脏器损伤,其中肝脏和神经系统病变最常见。其肝脏病理可表现为脂肪变性、炎症浸润、气球样变和Mallory小体生成等 。Mallory小体是细胞质中的嗜酸性包涵体,在蛋白质错误折叠和蛋白酶体功能受损等情况下可形成,主要成分为p62、泛素化蛋白和角蛋白K8/K18。前期研究表明激活自噬有助于Mallory小体的清除,但其具体机制尚不明确 。自噬是一种进化保守的维持细胞稳态的自我保护机制,已发现有四十余种ATG蛋白参与此过程 。其中半胱氨酸蛋白酶家族成员ATG4B可将未活化型的pro-LC3酶切为游离型的LC3-I,还可以将脂化型的LC3-II酶切回游离型LC3-I以循环利用。这两步酶切对于自噬体的形成至关重要,因此抑制ATG4B会导致自噬功能失活。ATG4B是否通过调节自噬而影响铜离子蓄积导致形成Mallory小体或清除尚不清楚。
该研究首先发现铜离子在体外蛋白水平可有效且特异性地抑制ATG4B蛋白的酶活性,并在细胞水平靶向ATG4B而抑制自噬;铜离子可引起p62和ATG4B在细胞水平形成聚集体,且在细胞和体外蛋白水平诱导ATG4B发生寡聚化,该寡聚化现象受到氧化还原调控。这些蛋白聚集现象与威尔森病中铜离子引起Mallory小体的生化特征相似,并且在威尔森病的细胞模型中,过表达ATG4B可一定程度逆转铜离子引起的自噬抑制以及相应的Mallory小体形成。综上,该研究发现了一种新的铜离子损伤机制,即铜离子靶向ATG4B而抑制自噬进而促进威尔森病中Mallory小体的生成并抑制其清除。该研究为全面认识铜离子与自噬的关系以及在威尔森病发病机制中的作用提供了新的实验证据。
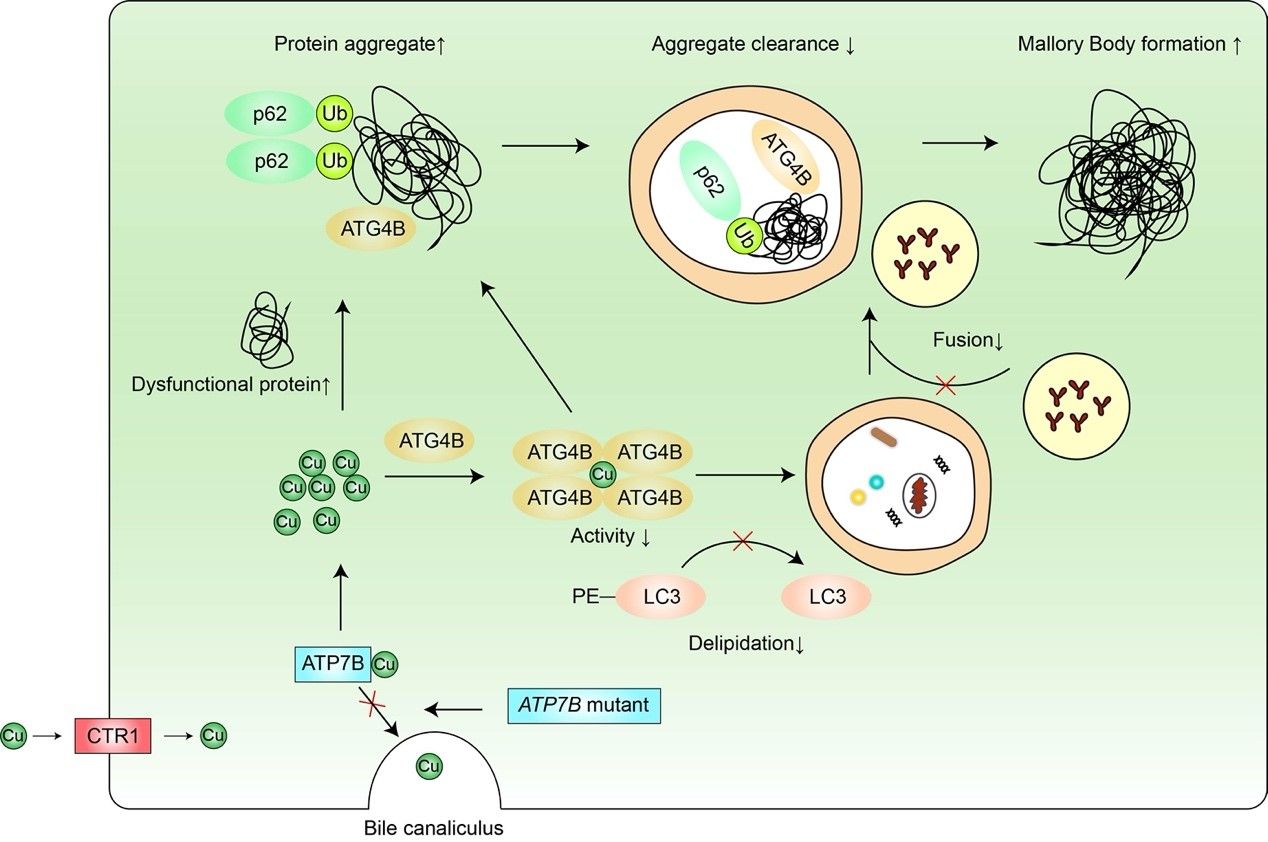
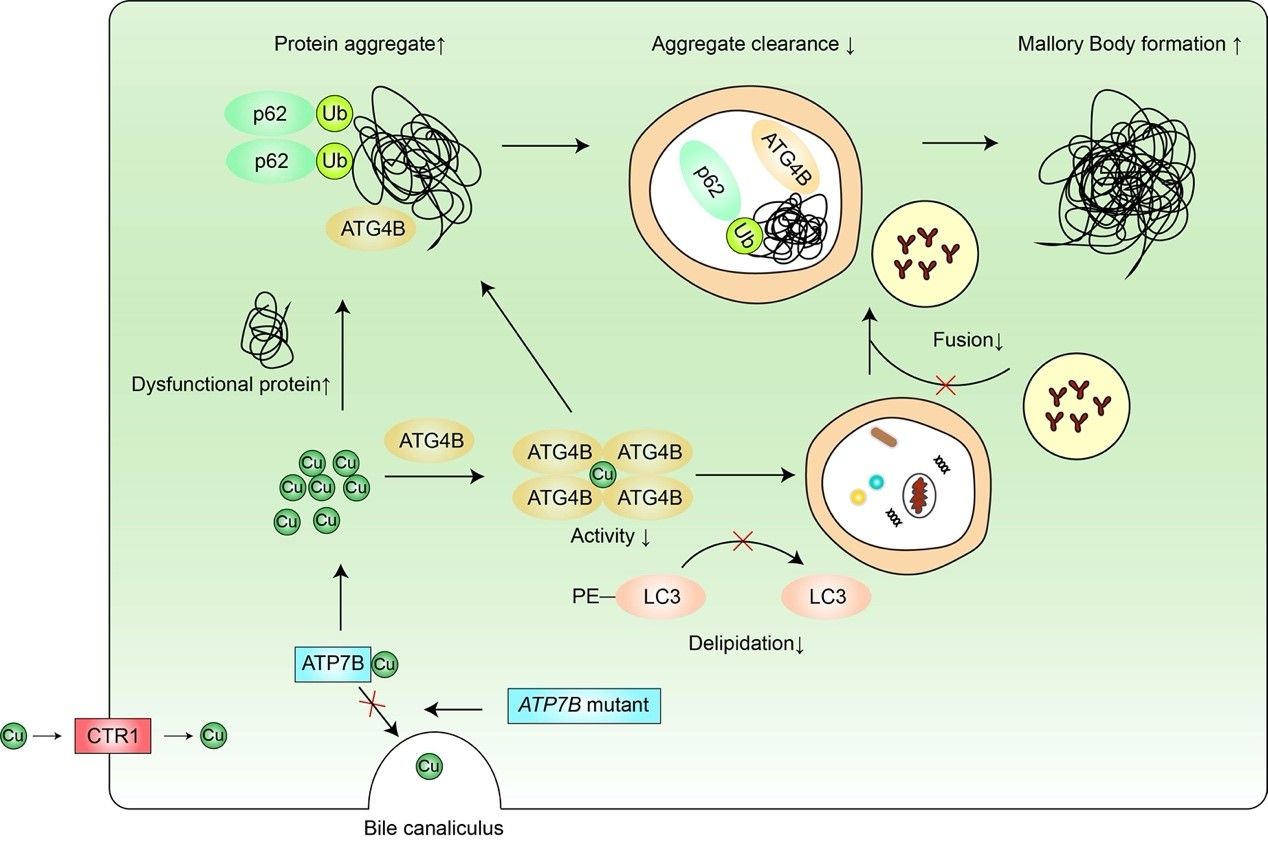
图1. ATG4B参与铜离子蓄积形成Mallory小体形成示意图
这项工作也是李民课题组前期发现在过氧化氢诱导条件下ATG4B氧化还原调控位点Cys292和Cys361研究工作的基础上( The protease activity of human ATG4B is regulated by reversible oxidative modification, Autophagy, 2020, 16: 1838-1850),新发现的一种铜离子介导的氧化还原调控模式。
中山大学药学院硕士生夏凡、博士后伏园园和硕士生谢华中为论文的共同第一作者,李民为通讯作者。中山大学药学院刘培庆团队和动物中心张薇团队为这项工作提供大力支持。该研究得到国家自然科学基金、广东省自然科学基金以及广东省手性分子与药物发现重点实验室的资助。